What is ALS?
ALS is also known as
Lou Gehrig’s Disease
Source & Credit: The ALS Association
Preface
Despite the serious nature of ALS, there are numerous people who live with ALS for many years – even decades – with a high quality of life. For some people they have a form of ALS that is very slowly progressing and for others they choose medical therapies and devices that help maintain mobility, nutrition and breathing. To learn more about the personal stories of people who are living fully, click here. As one man put it, “I’ve made ALS part of my life, not my whole life.”
What is ALS
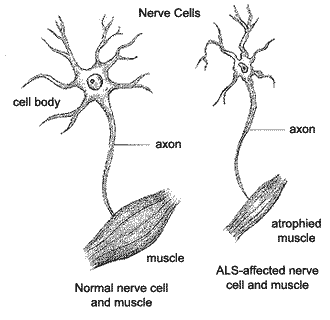
Amyotrophic Lateral Sclerosis (ALS), often referred to as "Lou Gehrig's disease," is a progressive neurodegenerative disease that affects nerve cells in the brain and the spinal cord. Motor neurons reach from the brain to the spinal cord and from the spinal cord to the muscles throughout the body. The progressive degeneration of the motor neurons in ALS eventually lead to their death. When the motor neurons die, the ability of the brain to initiate and control muscle movement is lost. With voluntary muscle action progressively affected, patients in the later stages of the disease may become totally paralyzed. Yet, through it all, for the vast majority of people, their minds remain unaffected.
A-myo-trophic comes from the Greek language. "A" means no or negative. "Myo" refers to muscle, and "Trophic" means nourishment---"No muscle nourishment." When a muscle has no nourishment, it "atrophies" or wastes away. "Lateral" identifies the areas in a person's spinal cord where portions of the nerve cells that signal and control the muscles are located. As this area degenerates it leads to scarring or hardening ("sclerosis") in the region.
As motor neurons degenerate, they can no longer send impulses to the muscle fibers that normally result in muscle movement. Early symptoms of ALS often include increasing muscle weakness, especially involving the arms and legs, speech, swallowing or breathing. When muscles no longer receive the messages from the motor neurons that they require to function, the muscles begin to atrophy (become smaller). Limbs begin to look "thinner" as muscle tissue atrophies.
What Types of Nerves Make Your Body Work Properly? |
(from Living with ALS, Manual 1: What's It All About?) |
The body has many kinds of nerves. There are those involved in the process of thinking, memory, and of detecting sensations (such as hot/cold, sharp/dull), and others for vision, hearing, and other bodily functions. The nerves that are affected when you have ALS are the motor neurons that provide voluntary movements and muscle power. Examples of voluntary movements are your making the effort to reach for the phone or step off a curb; these actions are controlled by the muscles in the arms and legs. |
The heart and the digestive system are also made of muscle but a different kind, and their movements are not under voluntary control. When your heart beats or a meal is digested, it all happens automatically. Therefore, the heart and digestive system are not involved in ALS. Breathing also may seem to be involuntary. Remember, though, while you cannot stop your heart, you can hold your breath - so be aware that ALS may eventually have an impact on breathing. |
Although the cause of ALS is not completely understood, the recent years have brought a wealth of new scientific understanding regarding the physiology of this disease.
While there is not a cure or treatment today that halts or reverses ALS, there is one FDA approved drug, Rilutek®, that modestly slows the progression of ALS as well as several other drugs in clinical trials that hold promise.
Importantly, there are significant devise and therapies that can manage the symptoms of ALS that help people maintain as much independence as possible and prolong survival. It is important to remember that ALS is a quite variable disease; no two people will have the same journey or experiences. There are medically documented cases of people in whom ALS ‘burns out,’ stops progressing or progresses at a very slow rate. No matter what your individual course or situation may be, The ALS Association and your medical team are here to help.
Initial Symptoms of the Disease
At the onset of ALS the symptoms may be so slight that they are frequently overlooked. With regard to the appearance of symptoms and the progression of the illness, the course of the disease may include the following:
- muscle weakness in one or more of the following: hands, arms, legs or the muscles of speech, swallowing or breathing
- twitching (fasciculation) and cramping of muscles, especially those in the hands and feet
- impairment of the use of the arms and legs
- "thick speech" and difficulty in projecting the voice
- in more advanced stages, shortness of breath, difficulty in breathing and swallowing
The initial symptoms of ALS can be
quite varied in different people. One person may experience tripping over carpet edges,
another person may have trouble lifting and a third person's early symptom may be slurred
speech. The rate at which ALS progresses can be quite variable from one person to another.
Although the mean survival time with ALS is three to five years, many people live five, ten
or more years. In a small number of people, ALS is known to remit or halt its progression,
though there is no scientific understanding as to how and why this happens. Symptoms can
begin in the muscles of speech, swallowing or in the hands, arms, legs or feet. Not all
people with ALS experience the same symptoms or the same sequences or patterns of
progression. But, progressive muscle weakness and paralysis are universally experienced.
Muscle weakness is a hallmark initial sign in ALS, occurring in approximately 60% of
patients. Early symptoms vary with each individual, but usually include tripping, dropping
things, abnormal fatigue of the arms and/or legs, slurred speech, muscle cramps and twitches
and/or uncontrollable periods of laughing or crying.
The hands and feet may be affected first, causing difficulty in lifting, walking or using
the hands for the activities of daily living such as dressing, washing and buttoning
clothes.
As the weakening and paralysis continue to spread to the muscles of the trunk of the body
the disease, eventually affects speech, swallowing, chewing and breathing. When the
breathing muscles become affected, ultimately, the patient will need permanent ventilatory
support in order to survive.
Since ALS attacks only motor neurons, the sense of sight, touch, hearing, taste and smell
are not affected. For many people, muscles of the eyes and bladder are generally not
affected.
For the vast majority of people, their mind and thoughts are not impaired and remain sharp
despite the progressive degenerating condition of the body.
Who Gets ALS
ALS is a disorder that affects the function of nerves and muscles. Based on U.S. population studies, a little over 5,600 people in the U.S. are diagnosed with ALS each year. (That's 15 new cases a day.) It is estimated that as many as 30,000 Americans have the disease at any given time. According to the ALS CARE Database, 60% of the people with ALS in the Database are men and 93% of patients in the Database are Caucasian.
Most people who develop ALS are
between the ages of 40 and 70, with an average age of 55 at the time of diagnosis. However,
cases of the disease do occur in persons in their twenties and thirties. Generally though,
ALS occurs in greater percentages as men and women grow older. ALS is 20% more common in men
than in women. However with increasing age, the incidence of ALS is more equal between men
and women.
There are several research studies – past and present – investigating possible risk factors
that may be associate with ALS. More work is needed to conclusively determine what genetics
and/or environment factors contribute to developing ALS.
Half of all people affected with ALS live at least three or more years after diagnosis. Twenty percent live five years or more; up to ten percent will live more than ten years.
There is some evidence that people with ALS are living longer, at least partially due to clinical management interventions, riluzole (Rilutek®) and possibly other compounds and drugs under investigation.
Forms of ALS
Three classifications of ALS have been described:
- Sporadic - the most common form of ALS in the United States - 90 to 95% of all cases.
- Familial - occurring more than once in a family lineage (genetic dominant inheritance) accounts for a very small number of cases in the United States - 5 to 10% of all cases.
- Guamanian - an extremely high incidence of ALS was observed in Guam and the Trust Territories of the Pacific in the 1950's.
The most common form of ALS in the United States is "sporadic" ALS. It may affect anyone, anywhere. "Familial" ALS (FALS) means the disease is inherited. Only about 5 to 10% of all ALS patients appear to have genetic or inherited form of ALS. In those families, there is a 50% chance each offspring will inherit the gene mutation and may develop the disease.
Diagnosing ALS
ALS is a very difficult disease to diagnose. To date, there is no one test or procedure to ultimately establish the diagnosis of ALS. It is through a clinical examination and series of diagnostic tests, often ruling out other diseases that mimic ALS, that a diagnosis can be established. A comprehensive diagnostic workup includes most, if not all, of the following procedures:
- electrodiagnostic tests including electomyography (EMG) and nerve conduction velocity (NCV)
- blood and urine studies including high resolution serum protein electrophoresis, thyroid and parathyroid hormone levels and 24 hour urine collection for heavy metals
- spinal tap
- x-rays, including magnetic resonance imaging (MRI)
- myelogram of cervical spine
- muscle and/or nerve biopsy
- thorough neurological examination
For more information on the importance of a second opinion, click here.
These tests are done at the discretion of the physician, usually based on the results of other diagnostic tests and the physical examination. There are several diseases that have some of the same symptoms as ALS and most of these conditions are treatable. It is for this reason that The ALS Association recommends that a person diagnosed with ALS seek a second opinion from an ALS "expert" - someone who diagnoses and treats many ALS patients and has training in this medial specialty. The ALS Association maintains a list of recognized experts in the field of ALS. See ALSA CentersSM, ALS Clinics and contact your local ALSA chapter or the National Office.
For more information, see Patient Services FAQ.
Genetic Testing for ALS
Writtin by Mara Gaudette, MS, Genetic Counselor
Formerly of Northwestern University
Updated November, 2004 by Lisa Dellefave
Q. Is ALS hereditary?
A. ALS is directly hereditary in only in a small percentage of families. The majority of patients with adult-onset ALS (90%) have no family history of ALS, and present as an isolated case. This is called sporadic ALS (SALS), and although there is likely a genetic predisposition involved, SALS is not directly inherited in a family. Rare exceptions are when familial ALS (FALS) is masked due to an incomplete family history, such as if the patient is adopted or the patient's parents died at ayoung age. The remaining10% of persons with ALS have a close second family member with ALS, which is referred to as familial ALS (FALS).
Currently the best tool to distinguish between SALS and FALS is the family history. A neurologist or genetic counselor will ask whether anyone else has ever been diagnosed with ALS, and if anyone else in the family had progressive walking or speech problems. If so, they will likely ask additional questions to see if the health problems were related to ALS or any number of other causes. They will also inquire about the ages that family members passed away to see if any close relatives passed away at a young age, meaning that a long health history is not available. It is very common to have limited information on one's family, but most families can still be reassured since the majority of instances of ALS are not hereditary. Older relatives are often good sources of family history information, and medical records can often be obtained with the help of a hospital's medical release form.
Q. How is FALS inherited?
A. To answer this question, it is
helpful to review background information on genetics
as it pertains to FALS. Every cell in the human body contains genes. Genes have many
functions. Some genes contribute to traits like eye and hair color while other genes are
responsible for making proteins that determine how our bodies circulate blood or send nerve
signals to muscles. When a gene is disrupted by a change in its sequence (called a gene
mutation), the gene cannot function correctly.
Genes are packaged in chromosomes. Chromosomes are present in pairs. The genes that reside
within chromosomes are therefore also present in pairs. In each chromosome pair, one
chromosome is inherited from the mother and one is inherited from the father. We have 23
pairs of chromosomes, giving us a total number of 46 chromosomes. The first 22 pairs are the
numbered chromosomes in which both males and females share them in common. Only the 23rd
pair differs between males and females since this pair is the sex chromosome where females
typically have two Xs and males have an X and a Y.
There are at least 3 different inheritance patterns for FALS. The most common inheritance
pattern for FALS is called autosomal dominant. Autosomal means that it is equally likely
that a female or male would inherit the gene mutation for FALS because the gene is located
on a numbered chromosome that both males and females share in common. Dominant refers to the
fact that a person only needs one gene to have a mutation coding for FALS to have an
increased risk for ALS. So someone who has FALS would have one gene with a mutation and one
gene witout a mutation. Therefore, a child born to someone who has FALS has a 50% chance to
inherit the FALS gene mutation and conversely, a 50% chance to not inherit the FALS gene
mutation. The 1 in 2, or 50% chance, comes from the fact that parents randomly pass on only
one member of their gene pair, so that either the gene with the mutation will be passed on
or the gene without the mutation will be passed on. Even though parents often feel
responsible for their children's health, they have no control over which gene they pass on,
just as their parent had no control which gene they passed onto their child. It is also
important to remember that inheriting the gene for FALS in no way guarantees that a person
will develop symptoms of ALS. If a child does not inherit the gene mutation for ALS, they
cannot pass it onto their children.
Q. Is there a genetic test for FALS?
A. Yes, although genetic testing is
still limited in FALS. Changes in one gene located on chromosome #21 and called superoxide
dismutase (SOD1) have been found in about 20% of families with FALS. The SOD1 gene is
composed of five regions called exons. If you think of your genetic material as a string of
letters that together make up a book of instructions for the human body, the SOD1 gene is
one chapter and composed of 5 different pages. SOD1's normal role is to detoxify substances
called free radicals, which can be harmful to cells. Changes in the SOD1 gene are thought to
create a new but still undefined function that is toxic to motor neurons. Most often, SOD1
changes are inherited in an autosomal dominant manner.
Of particular note is that the majority of families with FALS (80%) will not have a change
in their SOD1 gene and therefore, a normal SOD1 genetic test is not informative in a family
where an SOD1 change has not been identified. Although researchers are diligently looking
for other genes, at this time there is no genetic testing to offer non-SOD1 families.
Therefore, the determination that an individual has FALS is typically based on family
history rather than a genetic test.
Q. Does a genetic test diagnose ALS?
A. No. Since the vast majority of patients do not have the hereditary type of ALS, diagnosis of ALS is not determined by a genetic test. Instead, a neurologist makes the diagnosis after a review of a person's symptoms, a neurological exam, and results on nerve and muscle function tests. Clinically, FALS and SALS are basically identical.
Q. Who is appropriate for genetic testing?
A. Anyone who has symptoms of ALS in addition to a family history of ALS, such as a parent, grandparent, aunt or uncle, or a brother or sister. Additionally, if one's family history is unknown or a parent passed away at a young age, testing is also appropriate. However, only about 2% of all patients with ALS will have an SOD1 genetic change. Those patients with ALS without a family history can also be offered genetic testing but it is extremely important that it is offered in the context of genetic counseling or discussion with a neurologist about the implication of finding a mutation, as a mutation would mean the ALS is now hereditary in an apparently sporadic situation.
Q. What would the results of the genetic test tell me?
A. A positive test means that the
genetic cause of FALS has been identified. Researchers have developed a mouse model with the
same genetic change so that they can better understand how a change in the SOD1 gene can
lead to the symptoms of ALS. Currently, new therapies are being tried on this animal model
to slow or halt the progression of ALS. Although still in the distant future, gene therapy
to correct the genetic change is also being researched. A positive test does not change
medical treatment at this time and may or may not provide prognostic information. Even
though the inheritance may already be established by the family history, an individual may
feel furthered burdened by learning they carry a change in their SOD1 gene as concerns for
children resurface. Others prefer to have this knowledge and may feel comforted that there
is much research aimed specifically at ALS caused by changes in the SOD1 gene.
A negative test means only that the genetic cause of ALS has not been identified. However,
this does not rule out familial ALS since there are still other unidentified genes that
cause ALS in 80% of FALS families.
Q. If I have a family history of FALS, should I have a genetic test even if I don't have symptoms?
A. This situation is called
presymptomatic testing. The decision to have presymptomatic genetic testing is highly
personalized and often individuals in the same family will disagree whether to pursue it.
However, in order for the test to be meaningful, a genetic change in the SOD1 gene needs to
first be found in a family member affected with ALS. When an SOD1 change is not identified
in a symptomatic person, presymptomatic genetic testing is not available for other family
members, because the ALS is being caused by an unidentified gene, thus we cannot test for
it.
Benefits of presymptomatic genetic testing in ALS is limited by the absence of preventative
treatment, the inability to predict the age at which someone who is a gene carrier will get
ALS, or even that a gene carrier will definitely get ALS. Since both a negative or positive
presymptomatic test result in a known SOD1 family can have a great emotional impact, genetic
and psychological counseling is usually required before undergoing such testing (A
presymptomatic genetic testing protocol is typically followed). Individuals often consider
how the information that they did or did not inherit the predisposing gene would affect
their lives, who they would tell about the results, and how relationships may change
depending on the results.
Individuals who learn they do not carry the SOD1 change often feel great relief, although
they can sometimes wonder why they escaped while another family member did not. They may
regret past decisions made based on the presumed at risk status, or find it hard to let go
of that part of their identity. Learning that one does carry a predisposing gene, is usually
more difficult and that person may need ongoing professional support. Ambiguity is not
entirely erased as the question may change from do I carry the gene to when or will I get
symptoms? Commitment to friends and family may be strengthened. However, knowledge of the
testing by insurance companies or employers is a concern regarding future coverage. A
genetic counselor can further discuss the issues involved in presymptomatic testing.
Q. How is the genetic test done?
A. A blood sample is taken and sent to a specialized lab where the genetic material, also called DNA, is removed. Special laboratory techniques allow the SOD1 gene to be replicated and then tested. One form of testing is runnign the sample on a gel to generate a series of bands. If a genetic change is present, the bands will be in a different location compared to a control sample, which is known not to have a genetic change in the SOD1 gene. This method is called single strand conformation polymorphism or SSCP for short. Another method called sequencing may also be used to either initially test or confirm results. Sequencing is able to view the DNA on a finer scale by displaying the actual letters of the "instruction book" so that changes can be seen.
Q. How long does the genetic test take?
A. Because five different parts of
the SOD1 gene need to be looked at, the testing usually takes about 2-3 months. The cost is
about $300-500 depending on the clinical laboratory that is doing the testing.
If you have questions regarding genetic testing for ALS, you can contact:
Lisa Dellefave, MS, CGC
Genetic Counselor
Northwestern University Feinberg School of Medicine
(312) 503-0154
l-dellefave@northwestern.edu
This page was updated 12/2001.
Facts You Should Know About ALS
- The onset of ALS is insidious with muscle weakness or stiffness as early symptoms. Progression of weakness, wasting and paralysis of the muscles of the limbs and trunk as well as those that control vital functions such as speech, swallowing and later breathing generally follows.
- In most cases, mental faculties are not affected.
- ALS is not contagious.
- It is estimated that ALS is responsible for nearly two deaths per hundred thousand population annually. More people die every year of ALS than of Huntington's disease or multiple sclerosis and it occurs two-thirds as frequently as multiple sclerosis.
- Approximately 5,600 people in the U.S. are diagnosed with ALS each year. The incidence of ALS (two per 100,000 people) is five times higher than Huntington's disease and about equal to multiple sclerosis. It is estimated that as many as 30,000 Americans may have the disease at any given time.
- Although the life expectancy of an ALS patient averages about two to five years from the time of diagnosis, this disease is variable and many people live with quality for five years and more. More than half of all patients live more than three years after diagnosis.
- About twenty percent of people with ALS live five years or more and up to ten percent will survive more than ten years and five percent will live 20 years. There are people in whom ALS has stopped progressing and a small number of people in whom the symptoms of ALS reversed.
- ALS occurs throughout the world with no racial, ethnic or socioeconomic boundaries.
- ALS can strike anyone.
- Present treatment of ALS includes one drug, riluzole (Rilutek©) and is aimed at symptomatic relief, prevention of complications and maintenance of maximum optimal function and optimal quality of life. Most of this, in the later stages, requires substantial physical caregiving. Click here for more information on Rilutek.
- In 1991 a team of ALSA-funded researchers linked familial ALS to chromosome 21. In 1993 the research team identified a defective SOD1 gene on chromosome 21 as responsible for many cases of familial ALS. Further study indicated over 60 mutations (structural defects) in the SOD (superoxide dismutase) enzyme which alters the enzyme's ability to protect against free radical damage to motor neurons. These studies open possibilities for future therapies or strategies to effectively mediate both familial and sporadic ALS. But much more research on the SOD enzyme is needed. Also, researchers have not ruled out other gene involvement (on other chromosomes) in ALS.
- There can be significant costs for medical care, equipment and home health caregiving later in the disease. It is important to be knowledgeable about your health plan coverage and other programs for which your may be eligible, including SSA, Medicare, Medical and Veteran Affairs benefits.
- Rilutek®, the first treatment to
alter the course of ALS, was approved by the FDA in late 1995. This antiglutamate drug
was shown scientifically to prolong the life of persons with ALS by at least a few
months. More recent studies suggest Rilutek® slows the progress of ALS, allowing the
patient more time in the higher functioning states when their function is less affected
by ALS. Rilutek® is manufactured by Aventis Pharmaceuticals. There is a patient
Assistance Program that helps patients who qualify to receive the drug without charge.
Many private health plans cover the cost of Rilutek®. Contact your local
ALSA Chapter or the
National Office for the Patient Assistance
Program resource and more information about access to Rilutek®.
Reports from three separate patient databases described long range experience with Rilutek®. All three reports suggest a trend of increasing survival with Rilutek® over time. More studies that are double blind and controlled are needed to confirm these database observations. The trend appears to indicate that longer periods of time than those used in the Rilutek® clinical trials may be needed to see the long-term survival advantage of the drug. An interesting observation was that despite the fact that the Irish government provides Rilutek® free of charge to people in Ireland with ALS, only two-thirds of the patients registered in the Ireland national ALS database reported taking Rilutek.
Visitors
Count is per person no matter how many pages are visited!
Hits
Count is for ALL the hits each visitor makes!

prom dresses 2007 + size